
Europe
Beginning in the 1990s, European Union (EU) Medical Device Directives continually announced regulations related to medical devices. Aside from EU members, Australia, Switzerland, and other countries also consider EU’s Medical Device Directives as the reference for their medical device regulation. Through the Global Harmonization Task Force (GHTF), the content of medical device directives are used as globally harmonized directives for countries to refer to on related matters. Currently, Europe has the following directives related to medical devices. These directives include medical device classification rules, product certification routines, and requirements for quality systems and technical files.
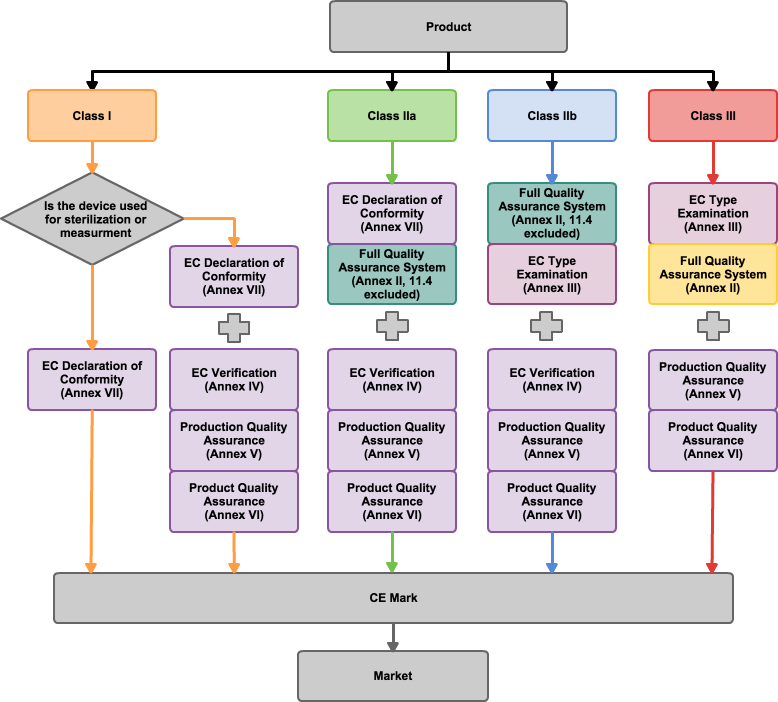
The European Commission is the executive body of the EU that is responsible for regulations and classifying medical devices into Class I, Class IIa, Class IIb, and Class III based on level of risk. The devices with higher risk must follow the strictest conformity assessments in order to receive the Conformité Européenne (CE) mark. CE marking is a mandatory legal requirement for most machinery, electric and electronic products, and many other products sold or put into service in Europe. The CE mark represents that the product is approved by the European Commission, meaning that it complies with their regulations. All medical devices that are intended to be sold an used in the EU requires the CE mark before entering the EU market.
There are two ways to obtain the CE Mark for medical devices. One is to be validated by notified bodies and obtain certificates from them. Another method is called “Declaration of Conformity”, in which firms can declare their products are safe with appropriate labeling and manufacturing information. However, the second method only works for medical devices with low risk. In cases with higher risk, the validation certifications by notified bodies is obligatory. Unlike the U.S. FDA system, which believes that applicants should possess a verified quality system, the European Commission requires validations on quality assurance systems for a CE marking submission. Quality assurance systems should construct and follow the corresponding directives to comply with CE standards. For CE marking, applicants need to submit a technical construction file (TCF) to provide scientific evidence and testing data to support their medical devices.
For foreign manufacturers not from the UN, a European Authorized Representative (E.A.R.) is required to apply for CE marking. A European Authorized Representative is a natural or legal person established in the European Economic Area ,EEA (including the EU and EFTA), who is explicitly designated by a foreign manufacturer to act on their behalf in carrying out certain tasks and regulations required for the applicable directives. The directives demand the E.A.R. to have a business with a registered address inside the EEA. The governments and authorities in the EEA have the righ to contact the E.A.R. at any time to verify that the manufacturers outside the EEA are meeting their obligations.
AcmeBiotechs provides services for applying for CE marking. We assist our clients with constructing quality assurance systems, preparing TCF, E.A.R, and going through the registration process. Our mission is to help clients to enter the EEA market without concerns about regulatory compliance.
- Active Implantable Medical Devices, AIMD, 90/76/EC
- Medical Devices Directives, MDD, 93/42/EEC
- In Vitro Diagnostic Medical Devices, IVDD, 98/79/EC
The European Commission is the executive body of the EU that is responsible for regulations and classifying medical devices into Class I, Class IIa, Class IIb, and Class III based on level of risk. The devices with higher risk must follow the strictest conformity assessments in order to receive the Conformité Européenne (CE) mark. CE marking is a mandatory legal requirement for most machinery, electric and electronic products, and many other products sold or put into service in Europe. The CE mark represents that the product is approved by the European Commission, meaning that it complies with their regulations. All medical devices that are intended to be sold an used in the EU requires the CE mark before entering the EU market.
There are two ways to obtain the CE Mark for medical devices. One is to be validated by notified bodies and obtain certificates from them. Another method is called “Declaration of Conformity”, in which firms can declare their products are safe with appropriate labeling and manufacturing information. However, the second method only works for medical devices with low risk. In cases with higher risk, the validation certifications by notified bodies is obligatory. Unlike the U.S. FDA system, which believes that applicants should possess a verified quality system, the European Commission requires validations on quality assurance systems for a CE marking submission. Quality assurance systems should construct and follow the corresponding directives to comply with CE standards. For CE marking, applicants need to submit a technical construction file (TCF) to provide scientific evidence and testing data to support their medical devices.
For foreign manufacturers not from the UN, a European Authorized Representative (E.A.R.) is required to apply for CE marking. A European Authorized Representative is a natural or legal person established in the European Economic Area ,EEA (including the EU and EFTA), who is explicitly designated by a foreign manufacturer to act on their behalf in carrying out certain tasks and regulations required for the applicable directives. The directives demand the E.A.R. to have a business with a registered address inside the EEA. The governments and authorities in the EEA have the righ to contact the E.A.R. at any time to verify that the manufacturers outside the EEA are meeting their obligations.
AcmeBiotechs provides services for applying for CE marking. We assist our clients with constructing quality assurance systems, preparing TCF, E.A.R, and going through the registration process. Our mission is to help clients to enter the EEA market without concerns about regulatory compliance.
CE marking Flowchart
